













Home
Our Mission
We educate leaders for Science, Medicine and Business!
We are Loschmidt Laboratories based at the Masaryk University and the International Clinical Research Center of St. Anne’s University Hospital Brno. Our laboratories are located at the University Campus Bohunice in Brno, Czech Republic.
Proteins are natural building blocks of all living organisms and participate in virtually every process within cells. Many natural proteins are enzymes that catalyse biochemical reactions and are essential for metabolism. Proteins can be found everywhere — they play a role in the development of medicines, in food and beverages processing, animal nutrition, textiles, household cleaning, fuels, and energy generation. And therefore the study of proteins is valuable for the whole society.
We conduct interdisciplinary research in the field of protein engineering. We wish to understand the structure-function relationships of proteins and improve their functionalities for biotechnologies. We study the mechanisms of Alzheimer’s disease and develop novel drugs for acute stroke. Our goal is to be recognized as one of the leading protein engineering groups in Europe. We will consistently strive to publish our findings in reputable scientific journals, develop software tools and lab-on-chips, and transfer results to practice.
The key components of our daily activities include collaboration between experimentalists and theoreticians, solidarity among the laboratory team, and mentoring of young colleagues — all while maintaining a friendly and creative working environment to honor the name of Jan Josef Loschmidt.
Profile Publications
EnzymeMiner 2.0: Advancing Automated Enzyme Discovery with Expansive Sequence Mining and Smart Property Analysis
Rosinska, M., Svobodova, L, Borko, S., Lacko, D., Planas-Iglesias, J., Marques, S. M., Kabourek, P., Liu, B., Pailozian, K., Damborsky, J., Mazurenko, S., Bednar, D., 2026, Nucleic Acids Research xx: gkag424.

Molecular Trick to Reverse SN2 Step in a Haloalkane Dehalogenase
Toul, M., Marques, S. M., Gao, T., Bernhardova, H., Vavra, O., Novakova, V., Damborsky, J., Bednar, D., Prokop, Z., Marek, M., 2026, Chem Catalysis 6: 101687.

FireProtDB 2.0: Large-Scale Manually Curated Database of the Protein Stability Data
Musil, M., Borko, S., Planas-Iglesias, J., Lacko, D., Rosinska, M., Kabourek, P., O Martins, L., Tataruch, M., Damborsky, J., Mazurenko, S., Bednar, D., 2026, Nucleic Acids Research 54: D409-D418.

Machine Learning Applied to Biocatalysis Research
Buller, R., Mazurenko, S., Yang, Y., 2025, Nature Communications 16: 1-5.

Quantum Computing for Faster Enzyme Discovery and Engineering
Damborsky, J., Kouba, P., Sivic, J., Vasina, M., Bednar, D., Mazurenko, S., 2025, Nature Catalysis 8: 872-880.

Structure Prediction and Computational Protein Design for Efficient Biocatalysts and Bioactive Proteins
Buller, R., Damborsky, J., Hilvert, D., Bornscheuer, U., 2025, Angewandte Chemie International Edition 64: e202421686.

Engineering Dehalogenase Enzymes Using Variational Autoencoder-Generated Latent Spaces and Microfluidics
Kohout, P., Vasina, M., Majerova, M., Novakova, V., Damborsky, J., Bednar, D., Marek, M., Prokop, Z., Mazurenko, S., 2025, JACS Au 5: 838-850.

Biochemical and Computational Characterization of Haloalkane Dehalogenase Variants Designed by Generative AI: Accelerating the SN2 Step
Gelfand, N., Orel, V., Cui, W., Damborsky, J., Li, C., Prokop, Z., Xie, W. J., Warshel, A., 2025, Journal of the American Chemical Society 147: 2747-2755.

Azobenzene-Based Photoswitchable Substrates for Advanced Mechanistic Studies of Model Haloalkane Dehalogenase Enzyme Family
Slanska, M., Stackova, L., Marques, S. M., Stacko, P., Martinek, M., Jilek, L., Toul, M., Damborsky, J., Bednar, D., Klan, P., Prokop, Z., 2024, ACS Catalysis 14: 11635-11645.

AggreProt: A Web Server for Predicting and Engineering Aggregation Prone Regions in Proteins
Planas-Iglesias, J., Borko, S., Swiatkowski, J., Elias, M., Havlasek, M., Salamon, O., Grakova, E., Kunka, A., Martinovic, T., Damborsky, J., Martinovic, J., Bednar, D., 2024, Nucleic Acids Research gkae420: 1-11.

CoVAMPnet: Comparative Markov State Analysis for Studying Effects of Drug Candidates on Disordered Biomolecules
Marques, S. M., Kouba, P., Legrand, A., Sedlar, J., Disson, L., Planas-Iglesias, J., Sanusi, Z., Kunka, A., Damborsky, J., Pajdla, T., Prokop, Z., Mazurenko, S., Sivic, J., Bednar, D., 2024, JACS Au 4: 2228-2245.

Illuminating the Mechanism and Allosteric Behavior of NanoLuc Luciferase
Nemergut, M., Pluskal, D., Horackova, J., Sustrova, T., Tulis, J., Barta, T., Baatallah, R., Gagnot, G., Novakova, V., Majerova, M., Sedlackova, K., Marques, S. M., Toul, M., Damborsky, J., Prokop, Z., Bednar, D., Janin, Y. L., Marek, M., 2023, Nature Communications 14: 7864.
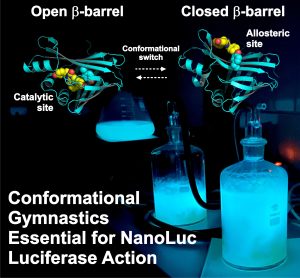
NanoLuc, a superior β-barrel fold luciferase, was engineered 10 years ago but the nature of its catalysis remains puzzling. In this work we reveal that imidazopyrazinone luciferins bind to an intra-barrel catalytic site but also to an allosteric site shaped on the enzyme surface. Structurally, binding to the allosteric site prevents simultaneous binding to the catalytic site, and vice versa, through concerted conformational changes. We demonstrate that restructuration of the allosteric site can boost the luminescent reaction in the remote active site. This information is critical to engineering the next-generation of ultrasensitive bioluminescent reporters.
Advancing Enzyme’s Stability and Catalytic Efficiency through Synergy of Force-Field Calculations, Evolutionary Analysis, and Machine Learning
Kunka, A., Marques, S. M., Havlasek, M., Vasina, M., Velatova, N., Cengelova, L., Kovar, D., Damborsky, J., Marek, M., Bednar, D., Prokop, Z., 2023, ACS Catalysis 13: 12506-12518.

Here, we compare different strategies for stabilizing enzymes, focusing on the hyperstable haloalkane dehalogenase DhaA115. Our results show that the automated protocols FireProt and PROSS outperformed rational design efforts in identifying multiple-point stabilizing mutations. The study also shows that manual curation of predicted mutations and machine learning algorithms can further improve stability and catalytic efficiency. The work resulted in three highly thermostable haloalkane dehalogenases with improved catalytic efficiencies.
In-depth Analysis of Biocatalysts by Microfluidics: An Emerging Source of Data for Machine Learning
Vasina, M., Kovar, D., Damborsky, J., Ding, Y., Yang, T., deMello, A., Mazurenko, S., Stavrakis, S., Prokop, Z., 2023, Biotechnology Advances 66: 108171.

In this review, we present the state-of-the-art, recent trends, and perspectives in applying microfluidic tools in the analysis of biocatalysts. We discuss the advantages and disadvantages of available technologies, their reproducibility and robustness, and their readiness for routine laboratory use. We also highlight the unexplored potential of microfluidics to leverage the power of machine learning for biocatalyst development.
Identification, Characterization, and Engineering of Glycosylation in Thrombolytics
Toul, M., Slonkova, V., Mican, J., Urminsky, A., Tomkova, M., Sedlak, E., Bednar, D., Damborsky, J., Hernychova, L., Prokop, Z., 2023, Biotechnology Advances 66: 108174.

Here we summarize current knowledge on computational and experimental identification of glycosylation sites and glycan identity, together with methods used for their reengineering. Practical examples from previous studies focus on modifying glycosylations in thrombolytics, e.g., alteplase, tenecteplase, reteplase, urokinase, saruplase, and desmoteplase. Collected clinical data on these glycoproteins demonstrate the great potential of the glycosylation engineering strategy for the developing a new generation of truly efficient and life-saving thrombolytic drugs.
Catalytic Mechanism for Renilla-type Luciferases
Schenkmayerova, A., Toul, M., Pluskal, D., Baatallah, R., Gagnot, G., Pinto, G. P., Santana, V. T., Stuchla, M., Neugebauer, P., Chaiyen, P., Damborsky, J., Bednar, D., Janin, Y. L., Prokop, Z., Marek, M., 2023, Nature Catalysis 6: 23-38.

Here we decipher Renilla-type catalysis through crystallographic, spectroscopic and computational experiments. Structures of ancestral and extant luciferases complexed with the substrate-like analogue azacoelenterazine or a reaction product were obtained, providing molecular snapshots of coelenterazine-to-coelenteramide oxidation. The results obtained also reveal structural features distinguishing flash-type from glow-type bioluminescence, providing insights that will guide the engineering of luciferases for diverse bioluminescent technologies.
Advanced Database Mining of Efficient Biocatalysts by Sequence and Structure Bioinformatics and Microfluidics
Vasina, M., Vanacek, P., Hon, J., Kovar, D., Faldynova, H., Kunka, A., Buryska, T., Badenhorst, C.P.S., Mazurenko, S., Bednar, D., Stavrakis, S., Bornscheuer, U.T., deMello, A., Damborsky, J., Prokop, Z., 2022, Chem Catalysis 2: 1-22.

We present a pipeline integrating sequence and structural bioinformatics with microfluidic enzymology to discover efficient and robust haloalkane dehalogenases. The bioinformatic part identified 2,905 putative dehalogenases and prioritized a “small-but-smart” set of 45 genes, yielding 40 active enzymes, 24 of which were biochemically characterized by microfluidic enzymology techniques. The obtained biocatalysts outperform the previously known wild-type and engineered dehalogenases. This strategy is applicable to other enzyme families, paving the way towards accelerating the identification of novel biocatalysts for industrial applications.
Mechanism-Guided Tunnel Engineering to Increase the Efficiency of a Flavin-Dependent Halogenase
Prakinee, K., Phintha, A., Visitsatthawong, S., Lawan, N., Sucharitakul, J., Kantiwiriyawanitch, C., Damborsky, J., Chitnumsub, P., van Pée, K.-H., Chaiyen, P., 2022, Nature Catalysis 5: 534-544.

Although flavin-dependent halogenases are attractive for C–H bond activation, their applications are limited due to low turnover and stability. Here we employed a mechanism-guided semi-rational approach to engineer the intermediate transfer tunnel connecting two active sites of tryptophan 6-halogenase Thal. This Thal-V82I variant generates less hypohalous acid leakage and possesses multiple catalytic improvements such as faster halogenation, broader substrate utilization, and greater thermostability and pH tolerance compared with the wild-type Thal.
CalFitter 2.0: Leveraging the Power of Singular Value Decomposition to Analyse Protein Thermostability
Kunka, A., Lacko, D., Stourac, J., Damborsky, J., Prokop, Z., Mazurenko, S., 2022, Nucleic Acid Research gkac249: 1-9.

Mechanism-Based Strategy for Optimizing HaloTag Protein Labeling
Marques, S. M., Slanska, M., Chmelova, K., Chaloupkova, R., Marek, M., Clark, S., Damborsky, J., Kool, E. T., Bednar, D., Prokop, Z., 2022, JACS Au 2: 1324−1337.

HaloTag labelling technology introduced unrivalled potential in protein chemistry and biology and it quickly became the dominant tagging technology. Surprisingly, despite the huge number of diverse applications and different available ligands, a single tagging enzyme is repeatedly used without considering the selection of other enzyme variants that are more suitable for a specific ligand. Our results show that natural dehalogenases provide high efficiency of the various ligand incorporation with no need for intensive optimization using directed evolution. Based on these findings, we propose a simple strategy for designing efficient HaloTag reactions, which consist of finding the optimal enzyme tag for specific ligand from a wide range of natural dehalogenases using easy-to-use in silico tools.
LoopGrafter: a Web Tool for Transplanting Dynamical Loops for Protein Engineering
Planas-Iglesias, J., Opaleny, F., Ulbrich, P., Stourac, J., Sanusi, Z., Pinto, G. P., Schenkmaye- rova, A., Byska, J., Damborsky, J., Kozlikova, B., Bednar, D., 2022, Nucleic Acid Research gkac249: 1-9.

Extended Mechanism of the Plasminogen Activator Staphylokinase Revealed by Global Kinetic Analysis: 1000-fold Higher Catalytic Activity than That of Clinically Used Alteplase
Toul, M., Nikitin, D., Marek, M., Damborsky, J., Prokop, Z., 2022, ACS Catalysis 12: 3807-3814.

We probed the complex kinetic mechanism of staphylokinase, an attractive thrombolytic drug candidate that is active in a multiprotein form with complex regulation. Using a modern global numerical approach, we found that the net value of its catalytic efficiency is 10,000-fold higher than previously determined by conventional methods limited by simplifying assumptions and approximations. We uncover the main catalytic bottleneck whose elimination might provide orders of magnitude more active alternative to the clinically used thrombolytic drug alteplase. Our findings pave the way for the development of a new generation of drugs effectively treating cardiovascular diseases.
Computational Enzyme Stabilization Can Affect Folding Energy Landscapes and Lead to Catalytically Enhanced Domain-Swapped Dimers
Markova, K., Kunka, A., Chmelova, K., Havlasek, M., Babkova, P., Marques, S. M., Vasina, M., Planas-Iglesias, J., Chaloupkova, R., Bednar, D., Prokop, Z., Damborsky, J., Marek, M., 2021, ACS Catalysis 11: 12864-12885.

Computer-encoded algorithms are increasingly employed to stabilize native proteins for research, biotechnologies, and biomedical applications. Methods such as FireProt are available to the research community as web services, making computer-aided design of multiple mutants extremely easy and widely accessible. This study reports important findings related to potential changes in the folding pathway and catalytic properties of designed hyperstable mutants. Structural analysis revealed that stabilizing mutations may activate cryptic hinge-loop regions and establish new secondary interfaces, leading to the formation of so-called domain-swapped dimer.
Engineering the Protein Dynamics of an Ancestral Luciferase
Schenkmayerova, A., Pinto, G. P., Toul, M., Marek, M., Hernychova, L., Planas-Iglesias, J., Daniel Liskova, V., Pluskal, D., Vasina, M., Emond, S., Dörr, M., Chaloupkova, R., Bednar, D., Prokop, Z., Hollfelder, F., Bornscheuer, U. T., Damborsky, J., 2021, Nature Communications 12: 3616.

We track the role of dynamics in evolution, starting from the evolvable and thermostable ancestral protein AncHLD-RLuc which catalyses both dehalogenase and luciferase reactions. Insertion-deletion backbone mutagenesis of AncHLD-RLuc challenged the scaffold dynamics. Screening for both activities reveals mutations localized in three distinct regions that lead to altered protein dynamics. Transplantation of a highly dynamic fragment leads to highly stable glow-type bioluminescence. The success of our approach suggests that a strategy comprising (i) constructing a stable and evolvable template, (ii) mapping functional regions by backbone mutagenesis, and (iii) transplantation of dynamic features, can lead to functionally innovative proteins.
Exploring Mechanism of Enzyme Catalysis by On-Chip Transient Kinetics Coupled with Global Data Analysis and Molecular Modeling
Hess, D., Dockalova, V., Kokkonen, P., Bednar, D., Damborsky, J., deMello, A., Prokop, Z., Stavrakis, S., 2021, Chem 7: 1-14.

We develop a droplet-based microfluidic platform for high-throughput acquisition of transient kinetic data over a range of substrate concentrations and temperatures. To demonstrate their utility, we measure the transient kinetics of three model enzymes, namely, b-galactosidase, horseradish peroxidase, and microperoxidase. Additionally, we conduct a complex kinetic and thermodynamic study of engineered variants of haloalkane dehalogenases. Datasets are globally analyzed and complemented by molecular dynamics simulations, providing new insights into the molecular basis of substrate specificity and the role of hydration-related entropy.
Decoding the Intricate Network of Molecular Interactions of a Hyperstable Engineered Biocatalyst
Markova, K., Chmelova, K., Marques, S. M., Carpentier, P., Bednar, D., Damborsky, J., Marek, M., 2020, Chemical Science 11: 11162–11178.

We report X-ray structures of a hyperstable engineered haloalkane dehalogenase (Tm = 73.5 °C), which highlight key thermostabilization effects. Unexpectedly, mutations toward bulky aromatic amino acids at the protein surface triggered long-distance (∼27 Å) backbone changes due to cooperative effects. These cooperative interactions produced an unprecedented double-lock system that dramatically reduced the volumes of enzyme access tunnels.
FireProtASR: A Web Server for Fully Automated Ancestral Sequence Reconstruction
Musil, M., Khan, R. T., Beier, A., Stourac, J., Konegger, H., Damborsky, J., Bednar, D., 2020, Briefings in Bioinformatics bbaa337: 1-11.

Ancestral sequence reconstruction is an engineering strategy applicable to the improvement of protein thermal stability and other characteristics. The FireProt-ASR provides users with one-stop-shop solution for ancestral sequence reconstruction. It fully automatizes all the steps: (i) search for the biologically relevant homolog sequences, (ii) reduction of the initial dataset, (iii) construction of the multiple-sequence alignment and the rooted phylogenetic tree, and (iv) reconstruction of the ancestral sequences. We employ a novel algorithm for the ancestral gaps reconstruction. FireProt-ASR provides easy-to-use user interface: http://loschmidt.chemi.muni.cz/fireprotasr/.
FireProtDB: Database of Manually Curated Protein Stability Data
Stourac, J., Dubrava, J., Musil, M., Horackova, J., Damborsky, J., Mazurenko, S., Bednar, D., 2020, Nucleic Acids Research 49: D319-D324.

The recent advances in machine learning facilitate the development of software tools for computational design of stable proteins. The accuracy of these tools strongly depends on the quality and amount of data used for training and testing. FireProtDB is a database of experimental thermostability data for single-point mutants. The database combines the published datasets, data extracted from the recent literature, and the data collected in our laboratory. Its user interface is designed to facilitate both types of the expected use: (i) the interactive explorations of individual entries and (ii) the construction of machine learning-friendly data sets: https://loschmidt.chemi.muni.cz/fireprotdb.
EnzymeMiner: Automated Mining of Soluble Enzymes with Diverse Structures, Catalytic Properties and Stabilities
Hon, J., Borko, S., Stourac J., Prokop, Z., Zendulka, J., Bednar D., Martinek, T., Damborsky, J., 2020, Nucleic Acids Research 48: W104–W109.

EnzymeMiner identifies putative members of enzyme families and facilitates the selection of promising targets for experiments. The server mines sequences that are likely to show the desired catalytic activity and prioritizes sequences heterologously expressible in a soluble form in Escherichia coli. EnzymeMiner reduces the time devoted to data gathering, multi-step analysis, sequence prioritization and selection from days to hours. EnzymeMiner is universally applicable to any enzyme family and provides an interactive and easy-to-use web interface freely available at https://loschmidt.chemi.muni.cz/enzymeminer/
Machine Learning in Enzyme Engineering
Mazurenko, S., Prokop., Z., Damborsky, J., 2019, ACS Catalysis 10: 1210-1223.

We analyse the state of the art in databases and machine learning methods used for training and validating predictors in enzyme engineering in this Perspective. We discuss current limitations and challenges which the community is facing and recent advancements in experimental and theoretical methods that have the potential to address those challenges. We also present our view on possible future directions for developing the applications to the design of efficient biocatalysts.
Caver Web 1.0: Identification of Tunnels and Channels in Proteins and Analysis of Ligand Transport
Stourac, J., Vavra, O., Kokkonen, P., Filipovic, J., Pinto, G., Brezovsky, J., Damborsky, J., Bednar, D., 2019, Nucleic Acid Research W1: W414–W422.

Caver Web 1.0 is a web server for comprehensive analysis of protein tunnels and channels, and study of the ligands’ transport through these transport pathways. Caver Web is the first interactive tool allowing both the analyses within a single graphical user interface. The tool is very fast (2-20 min per job) and is applicable even for virtual screening purposes. Its simple setup and comprehensive graphical user interface make the tool accessible for a broad scientific community. The server is freely available at https://loschmidt.chemi.muni.cz/caverweb.
Light-Emitting Dehalogenases: Reconstruction of Multifunctional Biocatalysts
Chaloupkova, R., Liskova, V., Toul, M., Markova, K., Sebestova, E., Hernychova, L., Marek, M., Pinto. G. P., Pluskal, D., Waterman, J., Prokop, Z., Damborsky, J., 2019, ACS Catalysis 9: 4810–4823.

To obtain structural insights into the emergence of new biological functions from catalytically promiscuous enzymes, we reconstructed an ancestor of catalytically distinct, but evolutionarily related, haloalkane dehalogenases (EC 3.8.1.5) and Renilla luciferase (EC 1.13.12.5). This ancestor has both hydrolase and monooxygenase activities. We demonstrate, that a single substitution next to the catalytic pentad enables the emergence of new activity at enzyme class-level. Ancestral reconstruction has a clear potential for obtaining multi-functional catalysts.
Exploring the Challenges of Computational Enzyme Design by Rebuilding the Active Site of a Dehalogenase
Jindal, G., Slanska, K., Kolev, V., Damborsky, J., Prokop, Z., Warshel, A., 2019, Proceedings of the National Academy of Sciences of the United States of America 116: 389–394.

The goal of rational computer-aided enzyme design is hampered by the lack of knowledge of the maximum possible rate enhancement. We address this problem by considering the enzyme DhlA, which is naturally adapted for the degradation of dihalogenated ethanes. Using empirical valence bond calculations, we determine the effect of finding mutations that reduce the catalysis and then introducing mutations that restore catalysis. One of our predicted cycles is confirmed experimentally, while the other attempt remains inconclusive. We believe that the proposed strategy provides a very powerful way of validating and refining approaches for computational enzyme design.
Molecular Gating of an Engineered Enzyme Captured in Real Time
Kokkonen, P., Sykora, J., Prokop, Z., Ghose, A., Bednar, D., Amaro, M., Beerens, K., Bidmanova, S., Slanska, M., Brezovsky, J., Damborsky, J., Hof, M., 2018, Journal of the American Chemical Society 140: 17999–18008.

Engineering dynamical molecular gates represents a widely applicable strategy for designing efficient biocatalysts. Here we analyzed the dynamics of a molecular gate artificially introduced into an access tunnel of the most efficient haloalkane dehalogenase using pre-steady-state kinetics, a single-molecule fluorescence spectroscopy and molecular dynamics. Photoinduced electron-transfer – fluorescence correlation spectroscopy (PET-FCS) has enabled real-time observation of molecular gating at single-molecule level with the rate constants (kon = 1822 s-1, koff = 60 s-1) corresponding well with those from the pre-steady-state kinetics (k-1 = 1100 s-1, k1 = 20 s-1).
Evolutionary Analysis is a Powerful Complement to Energy Calculations for Protein Stabilization
Beerens, K., Mazurenko, S., Kunka, A., Marques, S. M., Hansen, N., Musil, M., Chaloupkova, R., Waterman, J., Brezovsky, J., Bednar, D., Prokop, Z., Damborsky, J., 2018, ACS Catalysis 8: 9420−9428.

Stability is one of the most important characteristics of proteins and the role of computational approaches in modifying protein stability is rapidly expanding. Here we present a detailed mechanistic study of stabilizing mutations derived from the phylogenetic analysis. We explain why these highly beneficial mutations can be easily missed by widely used force-field calculations. A hybrid approach to protein stabilization – combining both energy calculation and evolutionary analysis – is freely available to the broad scientific community via the web server application FireProt: https://loschmidt.chemi.muni.cz/fireprot/.
CalFitter: A Web Server for Analysis of Protein Thermal Denaturation Data
Mazurenko, S., Stourac, J., Kunka, A., Nedejlkovic, S., Bednar, D., Prokop, Z., Damborsky, J., 2018, Nucleic Acids Research 46: W344-W349.

CalFitter web server is a unified platform for a comprehensive data fitting and an analysis of protein thermal denaturation data. The server allows simultaneous global data fitting using any combination of input data types and offers twelve protein unfolding pathway models to select from. The data fitting produces optimal parameters, their confidence intervals, and statistical information to define unfolding pathways. The server provides an interactive and easy-to-use interface that allows users to directly analyse input datasets: https://loschmidt.chemi.muni.cz/calfitter/.
HotSpot Wizard 3.0: Web Server for Automated Design of Mutations and Smart Libraries Based on Sequence Input Information
Sumbalova, L., Stourac, J., Martinek, T., Bednar, D., Damborsky, J., 2018, Nucleic Acids Research 46: W356-W362.

HotSpot Wizard is a web server for an automatic identification of ‘hot spots’ for the engineering of substrate specificity, activity or enantioselectivity of enzymes. The version 3.0 accepts the protein sequence as input data. The protein structure for the query sequence is obtained either from eight repositories of homology models or is modelled using Modeller and I-Tasser. A new module for the estimation of thermodynamic stabilities using the Rosetta suite has also been introduced which prevents destabilising mutations: http://loschmidt.chemi.muni.cz/hotspotwizard.
Exploration of Enzyme Diversity by Integrating Bioinformatics with Expression Analysis and Biochemical Characterization
Vanacek, P., Sebestova, E., Babkova, P., Bidmanova, S., Daniel, L., Dvorak, P., Stepankova, V., Chaloupkova, R., Brezovsky, J., Prokop, Z., Damborsky, J., 2018, ACS Catalysis 8: 2402–2412.

Millions of protein sequences are being discovered at an incredible pace, representing an inexhaustible source of biocatalysts. We present an integrated system for automated in silico screening and systematic characterization of diverse family members. The workflow consists of: (i) identification and computational characterization of relevant genes by sequence/structural bioinformatics, (ii) expression analysis and activity screening of selected proteins, and (iii) complete biochemical/biophysical characterization.
FireProt: Web Server for Automated Design of Thermostable Proteins
Musil, M., Stourac, J., Bendl, J., Brezovsky, J., Prokop, Z., Zendulka, J., Martinek, T., Bednar, D., Damborsky, J., 2017, Nucleic Acids Research 45: W393-W399.

FireProt is a web server for the automated design of multiple-point thermostable mutant proteins that combines structural and evolutionary information in its calculation core. FireProt utilizes sixteen tools and three protein engineering strategies for making reliable protein designs. The server is complemented with interactive, easy-to-use interface that allows users to directly analyze and optionally modify designed thermostable mutants. FireProt is freely available at https://loschmidt.chemi.muni.cz/fireprot.
Different Structural Origins of the Enantioselectivity of Haloalkane Dehalogenases toward Linear β-Haloalkanes: Open–Solvated versus Occluded–Desolvated Active Sites
Liskova, V., Stepankova, V., Bednar, D., Brezovsky, J., Prokop, Z., Chaloupkova, R., Damborsky, J., 2017, Angewandte Chemie International Edition 56: 4719-4723.

The enzymatic enantiodiscrimination of linear β-haloalkanes is difficult because the simple structures of the substrates prevent directional interactions. Herein we describe two distinct molecular mechanisms for the enantiodiscrimination of the β-haloalkane 2-bromopentane by haloalkane dehalogenases. Highly enantioselective DbjA has an open, solvent-accessible active site, whereas the engineered enzyme DhaA31 has an occluded and less solvated cavity but shows similar enantioselectivity. The enantioselectivity of DhaA31 arises from steric hindrance imposed by two specific substitutions rather than hydration as in DbjA.
Enzyme Tunnels and Gates as Relevant Targets in Drug Design
Marques, S. M., Daniel, L., Buryska, T., Prokop, Z., Brezovsky, J., Damborsky, J., 2016, Medicinal Research Reviews 37: 1095-1139.

We described a set of general concepts relating to the structural properties, function, and classification of enzyme tunnels and gates. We highlighted the potential of enzyme tunnels and gates as targets for the binding of small molecules, the different types of their binding, and the potential pharmacological benefits. Twelve examples of ligands bound to the tunnels and/or gates of clinically relevant enzymes were used to illustrate the different binding modes and to explain some new strategies for drug design.
Engineering a de Novo Transport Tunnel
Brezovsky, J., Babkova, P., Degtjarik, O., Fortova, A., Gora, A., Iermak, I., Rezacova, P., Dvorak, P., Kuta Smatanova, I., Prokop, Z., Chaloupkova, R., Damborsky, J., 2016, ACS Catalysis 6: 7597-7610.

We described the computational design and directed evolution of a de novo transport tunnel in a haloalkane dehalogenase. Mutants with a blocked native tunnel and a newly opened auxiliary tunnel in a distinct part of the structure showed dramatically modified properties. The mutants with blocked tunnels acquired specificity never observed with native family members: up to 32 times increased substrate inhibition and 17 times reduced catalytic rates. Opening of the auxiliary tunnel resulted in specificity and substrate inhibition similar to those of the native enzyme and the most proficient haloalkane dehalogenase reported to date.
PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions
Bendl, J., Musil, M., Stourac, J., Zendulka, J., Damborsky, J., Brezovsky, J., 2016, PLOS Computational Biology 12: e1004962.

We have developed a web server PredictSNP2 providing easy access to binary predictions and uniform confidence values for the five best-performing prediction tools and their consensus. These predictions are supplemented with information gathered from eight publicly available databases. PredictSNP2 extends the scope of genome analysis to the level of nucleotide substitutions that enables to identify disease-related variants within the whole genome.
HotSpot Wizard 2: Automated Design of Site-Specific Mutations and Smart Libraries in Protein Engineering
Bendl, J., Stourac, J., Sebestova, E., Vavra, O., Musil, M., Brezovsky, J., Damborsky, J., 2016, Nucleic Acids Research 44: W479-W487.

We developed HotSpot Wizard 2.0, a web server for automated identification of hot spots and design of smart libraries for engineering proteins’ stability, catalytic activity, substrate specificity and enantioselectivity. Compared to its predecessor, HotSpot Wizard 2.0 introduces several major improvements, extending the scope and quality of its analyses. It implements four different established protein engineering strategies, enabling the user to selectively target sites affecting the protein’s stability and catalytic properties. A new graphical interface provides an intuitive and comprehensive overview of the results of the analysis. The resulting pipeline of twenty integrated tools, including our in-house software Caver 3.0 for analysis of protein tunnels and channels, and three databases represents a unique one-stop solution that makes library design accessible even to users with no prior knowledge of bioinformatics.
FireProt: Energy- and Evolution-Based Computational Design of Thermostable Multiple-Point Mutants
Bednar, D., Beerens, K., Sebestova, E., Bendl, J., Khare, S., Chaloupkova, R., Prokop, Z., Brezovský, J., Baker, D., Damborsky, J., 2015, PLOS Computational Biology 11: e1004556.

FireProt is a robust computational strategy for predicting highly stable multiple-point mutants that combines energy- and evolution-based approaches with smart filtering to identify additive stabilizing mutations. We demonstrate that thermostability of the model enzymes can be substantially increased (ΔTm = 24°C and 21°C) by constructing and characterizing only a handful of multiple-point mutants. FireProt’s reliability and applicability was demonstrated by validating its predictions against 656 mutations from the ProTherm database. FireProt can be applied to any protein for which a tertiary structure and homologous sequences are available.
Site-specific Analysis of Protein Hydration Based on Unnatural Amino Acid Fluorescence
Amaro, M., Brezovsky, J., Kovacova, S., Sykora, J., Bednar, D., Nemec, V., Liskova, V., Kurumbang, N., Beerens, K., Chaloupkova, R., Paruch, K., Hof, M., Damborsky, J., 2015, Journal of the American Chemical Society 137: 4988-4992.

Hydration of proteins profoundly affects their functions. We describe a simple and general method for a site-specific analysis of protein hydration based on the in vivo incorporation of fluorescent unnatural amino acids and their analysis by steady-state fluorescence spectroscopy. Using this method, we investigate the hydration of functionally important regions of dehalogenases DhaA and DbjA. The experimental results are compared to findings from molecular dynamics simulations. Given the ongoing development of unnatural amino acids technology, this method could potentially be used to analyze hydration at specific sites in a wide range of proteins.
Dynamics and Hydration Explain Failed Functional Transformation in Dehalogenase Design
Sykora, J., Brezovsky, J., Koudelakova, T., Lahoda, M., Fortova, A., Chernovets, T., Chaloupkova, R., Stepankova, Prokop, Z., Kuta Smatanova, I., Hof, M., Damborsky, J., 2014, Nature Chemical Biology 10: 428-430.

We emphasize the importance of dynamics and hydration for enzymatic catalysis and protein design by transplanting the active site from a haloalkane dehalogenase with high enantioselectivity to nonselective dehalogenase. Protein crystallography confirms that the active site geometry of the redesigned dehalogenase matches that of the target, but its enantioselectivity remains low. Time-dependent fluorescence shifts and computer simulations revealed that dynamics and hydration at the tunnel mouth differ substantially between the redesigned and target dehalogenase.
PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations
Bendl J., Stourac J., Salanda O., Pavelka A., Wieben E.D., Zendulka J., Brezovsky J., Damborsky J., 2014, PLOS Computational Biology 10: e1003440.

We have constructed three independent datasets by removing duplicities, inconsistencies and mutations previously used in the training of evaluated tools. The benchmark dataset containing over 43,000 mutations was employed for the unbiased evaluation of eight established prediction tools: MAPP, nsSNPAnalyzer, PANTHER, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT and SNAP. The six best performing tools were combined into a consensus classifier PredictSNP, resulting into significantly improved prediction performance and robustness. The web server and the datasets are freely available to the academic community at https://loschmidt.chemi.muni.cz/predictsnp.
Gates of Enzymes
Gora, A., Brezovsky, J., Damborsky, J., 2013, Chemical Reviews 113: 5871–5923.

This review highlights the importance of gates in enzymes. The gates control substrate access to the active site and product release, restrict solvent access to specific protein regions, and synchronize processes occurring in distinct parts of the enzyme. Survey of 129 gates in 71 enzymes enabled a rigorous definition of gates and establishment of a new scheme for their classification. Gates were assigned to six distinct classes – wings, swinging doors, apertures, drawbridges, double drawbridges and shells. Presented are summary statistics describing the propensity of specific amino acid residues in particular gate classes. The proposed classification scheme provides guidance for the analysis and engineering of gates in biomolecular systems.
Engineering Enzyme Stability and Resistance to an Organic Cosolvent by Modification of Residues in the Access Tunnel
Koudelakova, T., Chaloupkova, R., Brezovsky, J., Prokop, Z., Sebestova, E., Hesseler, M., Khabiri, M., Plevaka, M., Kulik, D., Kuta Smatanova, I., Rezacova, P., Ettrich, R., Bornscheuer, U. T., Damborsky, J., 2013, Angewandte Chemie International Edition 52: 1959-1963.

Mutations targeting as few as four residues lining the access tunnel extended enzyme’s half-life in 40% dimethyl sulfoxide from minutes to weeks (4,000-fold) and increased its melting temperature by 19 °C. Protein crystallography and molecular dynamics revealed that the tunnel residue packing is a key determinant of protein stability and the active-site accessibility for co-solvent molecules (red dots). The broad applicability of this concept was verified by analyzing twenty six proteins with buried active sites from all six enzyme classes.
CAVER 3.0: A Tool for Analysis of Transport Pathways in Dynamic Protein Structures
Chovancova, E., Pavelka, A., Benes, P., Strnad, O., Brezovsky, J., Kozlikova, B., Gora, A., Sustr, V., Klvana, M., Medek, P., Biedermannova, L., Sochor, J., Damborsky, J., 2012, PLOS Computational Biology 8: e1002708.

Tunnels and channels facilitate the transport of small molecules, ions and water solvent in a large variety of proteins. CAVER is a software tool widely used for the identification and characterization of transport pathways in static macromolecular structures. A new version of CAVER was developed enabling automatic analysis of tunnels and channels in large ensembles of protein conformations. CAVER 3.0 implements new algorithms for the calculation and clustering of pathways. The software is freely available as a multiplatform command-line application at https://www.caver.cz.
Enantioselectivity of Haloalkane Dehalogenases and its Modulation by Surface Loop Engineering
Prokop, Z., Sato, Y., Brezovsky, J., Mozga, T., Chaloupkova, R., Koudelakova, T., Jerabek, P., Stepankova, V., Natsume, R., Leeuwen, J. G. E., Janssen, D. B., Florian, J., Nagata, Y., Senda, T., Damborsky, J., 2010, Angewandte Chemie International Edition 49: 6111-6115.

Engineering of the surface loop in haloalkane dehalogenases affects their enantiodiscrimination behavior. The temperature dependence of the enantioselectivity (lnE versus 1/T) of β-bromoalkanes by haloalkane dehalogenases is reversed (red data points) by deletion of the surface loop; the selectivity switches back when an additional single-point mutation is made. This behavior is not observed for α-bromoesters.
Redesigning Dehalogenase Access Tunnels as a Strategy for Degrading an Anthropogenic Substrate
Pavlova, M., Klvana, M., Chaloupkova, R., Banas, P., Otyepka, M., Wade, R., Nagata, Y., Damborsky, J., 2009, Nature Chemical Biology 5: 727-733.

Engineering enzymes to degrade anthropogenic compounds efficiently is challenging. We obtained Rhodococcus rhodochrous haloalkane dehalogenase mutants with up to 32-fold higher activity than wild type toward the toxic, recalcitrant anthropogenic compound 1,2,3-trichloropropane (TCP) using a new strategy. Key residues in access tunnels connecting the buried active site with bulk solvent by rational design were identified and randomized by directed evolution. The most active mutant has large aromatic residues at two out of three randomized positions and two positions modified by site-directed mutagenesis. These changes apparently enhance activity with TCP by decreasing accessibility of the active site for water molecules, thereby promoting activated complex formation.
HotSpot Wizard: a Web Server for Identification of Hot Spots in Protein Engineering
Pavelka, A., Chovancova, E., Damborsky, J., 2009, Nucleic Acids Research 37: W376-W383.

HotSpot Wizard is a web server for automatic identification of ‘hot spots’ for engineering of substrate specificity, activity or enantioselectivity of enzymes and for annotation of protein structures. The web server implements the protein engineering protocol, which targets evolutionarily variable amino acid positions located in the active site or lining the access tunnels. The ‘hot spots’ for mutagenesis are selected through the integration of structural, functional and evolutionary information obtained from: (i) the databases RCSB PDB, UniProt, PDBSWS, Catalytic Site Atlas and nr NCBI and (ii) the tools CASTp, CAVER, BLAST, CD-HIT, MUSCLE and Rate4Site. The HotSpot Wizard is freely available at https://loschmidt.chemi.muni.cz/hotspotwizard/.